Example data set
This example illustrates the input and output of the maxAlike web server. We use a multiple sequence alignment based on one of the multiz 44-way alignments of homo sapiens (hg18) and other vertebrate species. From the 19 sequences in this particular alignment, we want to reconstruct the most likely sequence for the dog (denoted as canFam2 in the phylogenetic tree).
Input
Sequence Alignment
The alignment in Stockholm format looks like this (only first columns are shown)
# STOCKHOLM 1.0 hg18 ATCATTATAACTGGCATTCTAATTGTAAAAAGAAACTTAAGGCAGATCTTTCTGGACAATAAC micMur1 ACCATTATAGCTGGGTTTCTAATTGTAAAAACAAACTCAAGCCAGATTTTCCTGGACAATAAC otoGar1 AGACTTTTAGCATGGATTCTAATTGGGAAAAGACACTCAAACCAGATCTTCCTGGACAATAAC tarSyr1 ATAATTATAGCTGCGATTATAATTATAAAAAGAAACTCAAGCCAGATCTCTCTGGACAATCAC calJac1 ATCATTATAACTGGCAACATAATTGTAAAAAGAAACGGCATGCAGAGCCTTCTGGTCAATAAC rheMac2 ATCATTATAACTGGCATTCTAATTGTCAAAAGAAACATAAGGCAGATCTTTCTGGACAATAAC ponAbe2 ATCATTATAACTGACATTCTAATCGTAAAAAGAAACTTAAGGCAGATCTTTCTGGACAATAAC gorGor1 ATCATTATAACTGGCATTCTAATTGTAAAAAGAAACTTAAGGCAGATCTTTCTGGACAATAAC panTro2 ATCATTATAACTGGCATTCTAATTGTAAAAAGAAACTTAAGGCAGATCTTTCTGGACAATAAC dipOrd1 ACCATTGTGGCAAGGAATATAACTGTAAAAAGAAACTCAAATCAGATTTTTCTGGAAAAAAAA ... speTri1 ATTATTATAGTTAGGATTATACTTACAAAAAGAGATTCAGGCCAGAGTTTTTCAGGAAAAGAA oryCun1 ATCGTGATCATTGGAATCCTAATTGTTAGAGAAGACACAACCCCGATCATTCTGGAAAAGAAC ochPri2 ACCATGACAGACAGAATTAAAATGATTAGAAAAGATACAGTCCGGATCATTCTGGACAGTAAT cavPor3 ACTATTGCATTTAGGAGTATAATTGCTCAGAGACACTCAAGTCAGATTTCTCCAGCAGATAAA turTru1 ACCGTCATAGCTAGGGTGATAATTATAAAACGAAATTCAAGCCAGATATTTCTGGAAAATAAC bosTau4 ACTGTTATAGCCAGGGTGATAGTTATAAAAAGGAACTCAAACCAGACCTTTCTGAGAAATAAC equCab2 ACTATTATAGCTAGGGTGATAATTATAAAAATAAACTCAAGCCAGATCTTTCTGGAAAATAAC loxAfr2 ACCATTATAGCTGGGTTGATAATTAATAAAAAAAACTCAAGCCAGATTTATCTGTGAAATAAC proCap1 ACTACCATAGCTGAACTGATAATTAATAAACAAAAAACAAGGTAGACTCATTTGTGAAATCTC //
Download the full alignment: example.stk.
Phylogenetic tree
The phylogenetic tree of all 44 species in Newick format looks like this:
((((((((((((((( ((hg18:0.006591,panTro2:0.006639):0.002184,gorGor1:0.009411):0.009942, ponAbe2:0.018342):0.014256,rheMac2:0.036199):0.021496, calJac1:0.066389):0.056911,tarSyr1:0.135169):0.011307, (micMur1:0.091452,otoGar1:0.128984):0.035463):0.015304, tupBel1:0.183583):0.004688,(((((mm9:0.083220,rn4:0.090564):0.196605, dipOrd1:0.209532):0.022555,cavPor3:0.223415):0.009828, speTri1:0.146894):0.025042, (oryCun1:0.116009,ochPri2:0.198295):0.100037):0.015355):0.020666, (((vicPac1:0.105252,(turTru1:0.064182,bosTau4:0.121911):0.025111):0.039691, ((equCab2:0.107726,(felCat3:0.097971,canFam2:0.100888):0.049486):0.006252, (myoLuc1:0.141155,pteVam1:0.111787):0.033187):0.004179):0.011699, (eriEur1:0.220580,sorAra1:0.266859):0.056117):0.021065):0.023276, (((loxAfr2:0.083775,proCap1:0.152633):0.026190,echTel1:0.240221):0.049905, (dasNov2:0.115179,choHof1:0.096272):0.052373):0.006713):0.232748, monDom4:0.325899):0.072430,ornAna1:0.453916):0.109903, ((galGal3:0.166386,taeGut1:0.170717):0.199763, anoCar1:0.509545):0.108130):0.166150,xenTro2:0.852482):0.300396, (((tetNig1:0.224774,fr2:0.205294):0.191836, (gasAcu1:0.313967,oryLat2:0.478451):0.058404):0.322824, danRer5:0.731166):0.155214):0.511293,petMar1:0.511293);
Download the tree file: example.ph.
Note that the names in the tree must match the names in the alignment file.
Target species
Since we want to reconstruct the most likely sequence of the dog, we use canFam2 as the target name.
Output
Nucleotide probabilities
The main output file contains one line for each alignment column (only first 20 lines are shown).
0 pair=-1 max_mu=0 IC=2.32193 A=1 G=0 C=0 T=0 -=0 1 pair=-1 max_mu=2.11763 IC=1.16445 A=0.0674266 G=0.0437362 C=0.753695 T=0.135142 -=0 2 pair=-1 max_mu=3.56614 IC=0.678139 A=0.110643 G=0.0717678 C=0.508599 T=0.30899 -=0 3 pair=-1 max_mu=2.18847 IC=0.943766 A=0.644017 G=0.240088 C=0.051334 T=0.0645603 -=0 4 pair=-1 max_mu=0.457851 IC=1.91297 A=0.0171434 G=0.01112 C=0.0305824 T=0.941154 -=0 5 pair=-1 max_mu=1.49823 IC=1.37601 A=0.0529735 G=0.0343613 C=0.0931442 T=0.819521 -=0 6 pair=-1 max_mu=1.68144 IC=1.2241 A=0.770531 G=0.13305 C=0.0427074 T=0.0537113 -=0 7 pair=-1 max_mu=0.935887 IC=1.63752 A=0.0341254 G=0.0221354 C=0.0587983 T=0.884941 -=0 8 pair=-1 max_mu=1.27804 IC=1.37449 A=0.81497 G=0.108736 C=0.0337935 T=0.0425006 -=0 9 pair=-1 max_mu=1.04112 IC=1.58642 A=0.0936975 G=0.864318 C=0.0185963 T=0.0233878 -=0 10 pair=-1 max_mu=1.63799 IC=1.32655 A=0.0530898 G=0.0344366 C=0.803132 T=0.109341 -=0 11 pair=-1 max_mu=2.18761 IC=1.16107 A=0.0745705 G=0.04837 C=0.119688 T=0.757371 -=0 12 pair=-1 max_mu=2.34882 IC=0.824536 A=0.520819 G=0.362756 C=0.0515687 T=0.0648559 -=0 13 pair=-1 max_mu=0.933621 IC=1.63947 A=0.0847809 G=0.877595 C=0.0166649 T=0.0209587 -=0 14 pair=-1 max_mu=1.07897 IC=1.56838 A=0.096802 G=0.859678 C=0.0192765 T=0.0242432 -=0 15 pair=-1 max_mu=2.67404 IC=0.91067 A=0.268895 G=0.61654 C=0.050638 T=0.0639265 -=0 16 pair=-1 max_mu=1.59647 IC=1.35679 A=0.056158 G=0.0364268 C=0.0923249 T=0.81509 -=0 17 pair=-1 max_mu=1.27631 IC=1.47305 A=0.112811 G=0.834471 C=0.0229354 T=0.0297822 -=0 18 pair=-1 max_mu=2.86178 IC=0.919825 A=0.664303 G=0.185951 C=0.0671282 T=0.0826174 -=0 19 pair=-1 max_mu=0.47544 IC=1.90122 A=0.0177845 G=0.0115359 C=0.0316836 T=0.938996 -=0
Download the full output file: example.out
Sequence logo
The probabilities are shown in the sequence logo:
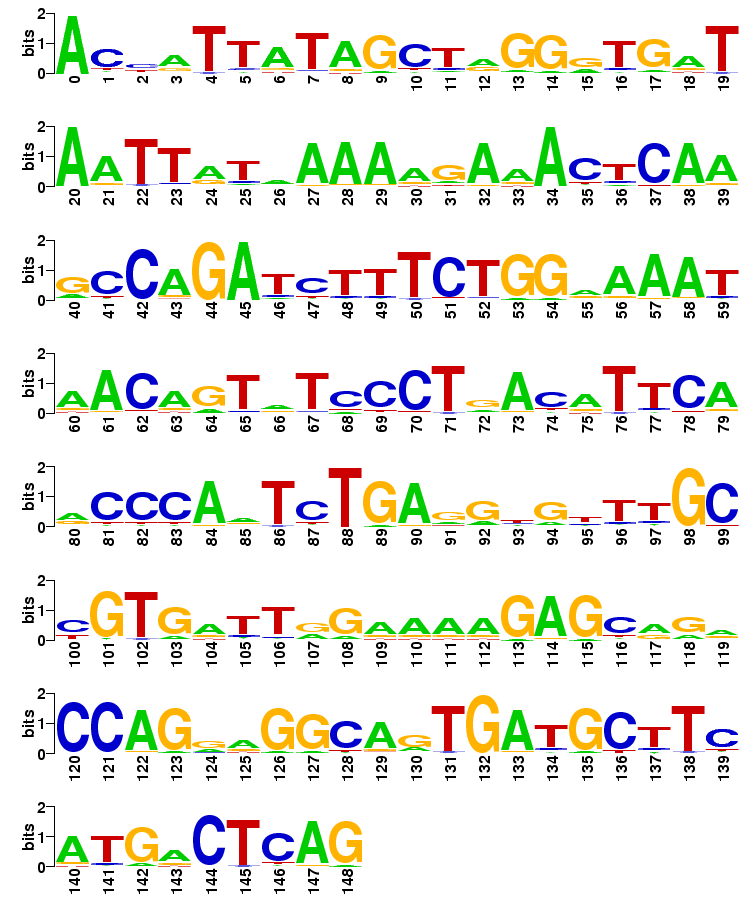
Consensus sequences
Predicted consensus sequence, with a probability cutoff 0.5 at each site:
>canFam2 predicted consensus sequence with probability cutoff 0.5
ACCATTATAGCTAGGGTGATAATTATAAAAAGAAACTCAAGCCAGATCTTTCTGGAAAATAACAGTATCCCTGACATTCA
ACCCAATCTGAGGTGTTTGCCGTGATTGGAAAAGAGCAGACCAGGAGGCAGTGATGCTTCATGACTCAG
Predicted consensus sequence, without cutoff:
>canFam2 predicted consensus sequence
ACCATTATAGCTAGGGTGATAATTATAAAAAGAAACTCAAGCCAGATCTTTCTGGAAAATAACAGTATCCCTGACATTCA
ACCCAATCTGAGGTGTTTGCCGTGATTGGAAAAGAGCAGACCAGGAGGCAGTGATGCTTCATGACTCAG
Conserved Elements
Subsequences having a window length of 10nt with average information content above 1.5:
![]()
![]()
![]()
![]()
![]()
![]()