|
|
|
About these examplesThese have been written as an introduction to the programs, their output, as well as the column format. More detailed description can be found on the man pages.
The basicsGenerating a col fileTo generate a col file, we can start with a number of different sequence files. One possibility is to use fasta format ad the col2fasta program. Another possibility is to use the following txt format:pairing_mask --3---311111111----222222-----111111111--2222222----- orga -gAcugUcugUCGAUgauu-AAcGuac--cAUCGAccgug-aCaUUuagua-- orgb agAccgUcg-UCGAUGauu-AAcGuaca-CAUCGA-cgug-aCaUUuagua-- orgc cgAcacUcuguCCAUgauu-AAcGuac--aAUCGaacggu-aCaUUuaguacc orgd -gAcchUgcguCGAUgauu-AAcGuac-uaAUCGaacgug-aCaUUuagua--In this format the structure is given by a pairing mask as the first sequence. The bases which form pairs are given as capital letters in the RNA sequences. Pairs are formed between bases that has the same symbols in the pairing mask. From this format, a col file can be made by writing:
txt2col example.txt > example.col Now example.col contains the col file. The start of the resulting col file looks like this: ; Generated by txt2col ; ======================================================================== ; TYPE pairingmask ; COL 1 label ; COL 2 residue ; COL 3 seqpos ; ENTRY pairing_mask ; ---------- M - 1 M - 2 M 3 3 M - 4 M - 5 M - 6 M 3 7 M 1 8 M 1 9 M 1 10 M 1 11 M 1 12The file starts with a header that describes where the file came from. This one was generated by txt2col. This area of the file could also contain information written by the person who made the file. In a database, this could be a reference to the article describing the article, which version it is etc. The header is ended by the line of equality sign. Notice how all lines starting with semicolons are comments. The first entry of the file has the type pairingmask which describes RNA structure. It was the -m option to txt2col that made the program include the pairing mask as an entry. It is not this entry that specifies the structure of the following RNA sequences, this information is kept in each entry as shown below. This means that the pairing mask is not necessary, but it is only kept as a reference. The first column in this entry is a label that describes what is in each position. Here it is all M's for pairingmask. The seconde column called residue contains the symbols of the sequences. The third column contains the position numbers in the sequence. The next entry in the col file is the first real sequence, that starts like this: ; TYPE RNA ; COL 1 label ; COL 2 residue ; COL 3 seqpos ; COL 4 alignpos ; COL 5 align_bp ; ENTRY orga ; ---------- G - . 1 . N g 1 2 . N A 2 3 7 N c 3 4 . N u 4 5 . N g 5 6 . N U 6 7 3 N c 7 8 . N u 8 9 . N g 9 10 . N U 10 11 35 N C 11 12 34This is of type RNA. The first column is again a label, here all the nucleotides have N in this column, while gaps have G's. Column two contains the sequence symbols and column three contains the sequence positions. The fourth column has positions relative to the alignment. The fifth column is called align_bp, for align basepair. This has the secondary structure of the RNA, specified as pairs relative to the alignpos column. A dot in the column means that the nucleotide is unpaired. The entire col file can be found here.
Making postscript alignmentsTo make a nice looking figure of an alignment, the program col2psalign is useful:
col2psalign --figure example.col > example_1_1.ps This makes a postscript file that looks like this:
The figure can also be made to look like this:
This is done by the command:
col2psalign --figure --space --textwidth=32 example.col > example_1_2.ps The --space option inserts spaces for every ten positions in the alignment. The --textwidth option is used to specify how wide the alignment should be. The value of textwidth is the number of characters after the sequence name, including the spaces input by the --space option. Another figure:
This was made with the command:
col2psalign --figure --range=10-20,30-40 example.col > example_1_3.ps This is useful to illustrate interesting parts of the alignment.
Checking RNA structuresStrange nucleotidesA col file can be checked for non-standard nucleotides. The program unknown can do this:
unknown example.col | col2psalign --figure > example_2_1.ps Here, the col file is altered by stdpair and then sent to col2psalign to make a figure that looks like this:
This shows that organism orgd contain an h is it sequence. This could be an error. Let us change the h to a gap and call the file example2.txt. From this a col file is generated:
txt2col -m example2.txt > example2.col
Strange base pairsA col file with RNA sequences can be checked for non-standard base pairs. This is done with the program stdpair:
stdpair --color example2.col | col2psalign --figure > example_2_2.ps It looks like this:
This has highlighted a C-C pair in the orgc sequence. It was the --color option that made the program color the nucleotides. Without this option, the strange pair would have been removed (notice that the change happens only in the align_bp column, it does not change the nucleotides to lowercase letters). The sequence orgb has highlighted nucleotides as well. This basepair looks fine, but no pairing mask tells txtcol which columns should pair in these positions. txt2col sets the bases to pair with themselves, to show that there is no pairing mask in these positions. To see this in the col file, the programs grepcol and greppos are useful:
grepcol --range=orgb example2.col | greppos --range=14-32 > example_2_1.col This gives the following result: ; Generated by txt2col ; ; 'grepcol --range=orgb' was run on this file ; ; 'greppos --range=14-32' was run on this file ; ======================================================================== ; TYPE RNA ; COL 1 label ; COL 2 residue ; COL 3 seqpos ; COL 4 alignpos ; COL 5 align_bp ; ENTRY orgb ; ---------- N A 13 14 32 N U 14 15 31 N G 15 16 16 N a 16 17 . N u 17 18 . N u 18 19 . G - . 20 . N A 19 21 46 N A 20 22 45 N c 21 23 . N G 22 24 43 N u 23 25 . N a 24 26 . N c 25 27 . N a 26 28 . G - . 29 . N C 27 30 30 N A 28 31 15 N U 29 32 14 ; **********Notice that alignpos 16 and 30 has themselves as pairs. This could be errors in the txt file and could be correct to give a new file, example3.txt: pairing_mask --3---3111111111---222222----1111111111--2222222----- orga -gAcugUcugUCGAUgauu-AAcGuac--cAUCGAccgug-aCaUUuagua-- orgb agAccgUcg-UCGAUGauu-AAcGuaca-CAUCGA-cgug-aCaUUuagua-- orgc cgAcacUcuguCcAUgauu-AAcGuac--aAUcGaacggu-aCaUUuaguacc orgd -gAcc-UgcguCGAUgauu-AAcGuac-uaAUCGaacgug-aCaUUuagua--From this, a col file is generated:
txt2col -m example3.txt > example3.col
Stems that can be extendedTo illustrate which stems that could be extended, the extendstem program is useful:
extendstem --gupair --color example3.col | col2psalign --figure > example_2_3.ps This command colors the positions that could be paired to extend existing stems. The --gupair option makes the program treat G-U pairs like the standard A-U and G-C pairs.
This could lead to the following text file, example4.txt: pairing_mask -33---3311111111---22222222--1111111111222222222----- orga -GAcugUCuGUCGAUGauu-AAcGUac--CAUCGACcgug-ACaUUuagua-- orgb agAccgUc-GUCGAUGauu-AAcGUaca-CAUCGAC-gug-ACaUUuagua-- orgc cGAcacUCugUCcAUgauu-AAcGUAC--aAUcGAacg-GUACaUUuaguacc orgd -gAcc-UgcgUCGAUgauu-AAcGUac-uaAUCGAacgug-ACaUUuagua--From this, yet another col file is generated:
txt2col -m example4.txt > example4.col To give an impression of the result of the changes made to example.txt, the following commands can be run:
txt2col -m example.txt | unknown | stdpair --color | extendstem --color | col2psalign --figure > example_2_4.ps
txt2col -m example4.txt | unknown | stdpair --color | extendstem --color | col2psalign --figure > example_2_5.ps This shows that many commands can be combined. The results are shown here: Before:
After:
The nucleotide that is colored cyan can be part of two stem extensions. This is the reason that its structure was not changed. Other programsMaking fasta filesMaking fasta files from col files:
col2fasta example3.col > example.fasta This gives: >pairing_mask --3---3111111111---222222----1111111111--2222222----- >orga -GACUGUCUGUCGAUGAUU-AACGUAC--CAUCGACCGUG-ACAUUUAGUA-- >orgb AGACCGUCG-UCGAUGAUU-AACGUACA-CAUCGA-CGUG-ACAUUUAGUA-- >orgc CGACACUCUGUCCAUGAUU-AACGUAC--AAUCGAACGGU-ACAUUUAGUACC >orgd -GACC-UGCGUCGAUGAUU-AACGUAC-UAAUCGAACGUG-ACAUUUAGUA--If the gaps are not wanted, use:
nogap example3.col | col2fasta > example.nogap.fasta To give: >pairing_mask --3---3111111111---222222----1111111111--2222222----- >orga GACUGUCUGUCGAUGAUUAACGUACCAUCGACCGUGACAUUUAGUA >orgb AGACCGUCGUCGAUGAUUAACGUACACAUCGACGUGACAUUUAGUA >orgc CGACACUCUGUCCAUGAUUAACGUACAAUCGAACGGUACAUUUAGUACC >orgd GACCUGCGUCGAUGAUUAACGUACUAAUCGAACGUGACAUUUAGUAThe pairing mask is probably not wanted in this case, and txt2col should be used without the -m option:
txt2col example3.txt | nogap | col2fasta > example2.nogap.fasta To give: >orga GACUGUCUGUCGAUGAUUAACGUACCAUCGACCGUGACAUUUAGUA >orgb AGACCGUCGUCGAUGAUUAACGUACACAUCGACGUGACAUUUAGUA >orgc CGACACUCUGUCCAUGAUUAACGUACAAUCGAACGGUACAUUUAGUACC >orgd GACCUGCGUCGAUGAUUAACGUACUAAUCGAACGUGACAUUUAGUA Showing structureThe secondary structure of RNA can be shown using the addparen program. This adds a sequence of parentheses after each RNA sequence:
addparen example.col | col2psalign --figure > example_3_1.ps This makes the alignment look like this:
The matching parentheses shows which nucleotides form pairs. Notice that two types of parentheses are used, because these structures have pseudoknots. The positions that pairs with themselves (see above) are indicated with an `x'. If a program like stdpair is run without the --color option, the structure is changed instead of colored. When pairs are removed, the letters are not changed to lower case, since the case of the letter were only used by the txt2col to find the structure. From that point on, the structure was given in the align_bp column of the col file:
stdpair example.col | addparen | col2psalign --figure > example_3_2.ps This makes the alignment look like this:
Text alignmentsSometimes, it can be useful to make text alignments that are easy to look at. This can be done with the col2txtalign program:
col2txtalign example4.col The output from this looks like this: 1 53 pairing_ma -33---3311111111---22222222--1111111111222222222----- orga -GAcugUCuGUCGAUGauu-AAcGUac--CAUCGACcgug-ACaUUuagua-- orgb agAccgUc-GUCGAUGauu-AAcGUaca-CAUCGAC-gug-ACaUUuagua-- orgc cGAcacUCugUCcAUgauu-AAcGUAC--aAUcGAacg-GUACaUUuaguacc orgd -gAcc-UgcgUCGAUgauu-AAcGUac-uaAUCGAacgug-ACaUUuagua--The relevant commands from col2psalign can also be used for col2txtalign:
col2txtalign --space --textwidth=33 --namewidth=15 example4.col This gives the following output: 1 30 pairing_mask -33---3311 111111---2 2222222--1 orga -GAcugUCuG UCGAUGauu- AAcGUac--C orgb agAccgUc-G UCGAUGauu- AAcGUaca-C orgc cGAcacUCug UCcAUgauu- AAcGUAC--a orgd -gAcc-Ugcg UCGAUgauu- AAcGUac-ua 31 53 pairing_mask 1111111112 22222222-- --- orga AUCGACcgug -ACaUUuagu a-- orgb AUCGAC-gug -ACaUUuagu a-- orgc AUcGAacg-G UACaUUuagu acc orgd AUCGAacgug -ACaUUuagu a-- Comments, questions, etc., email
gorodkin@rth.dk. |
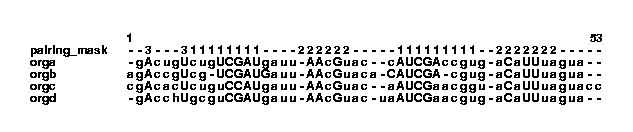

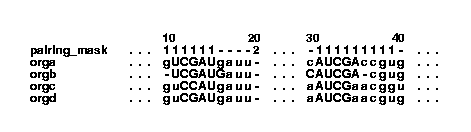
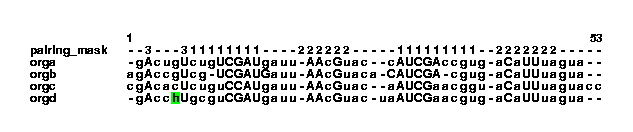
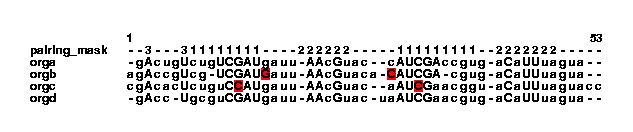
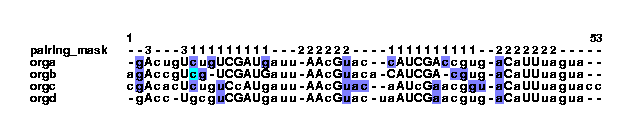
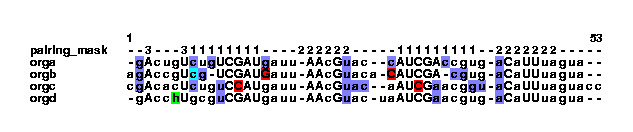
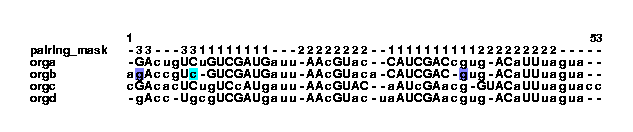


| Last updated March 26th, 2007 by Jan Gorodkin |