Help
Contact
Please contact Peter Menzel (ptr@rth.dk) for any technical issues or general questions about the software.
Download
The sources for maxAlike can be downloaded here (Linux).
Introduction
The maxAlike algorithm aims at reconstructing a nucleotide sequence in a target species, based on a multiple sequence alignment and a phylogenetic tree of homologous sequences from other species. For each alignment position the probabilities of occurrence for each nucleotide are computed, considering the phylogenetic position of the target species in the tree.
The computation is performed by a maximum likelihood algorithm. The resulting probabilities can for instance be used to construct homology search models (e.g. based on position weight matrices) or to derive short sequences for designing primers for yet to be sequenced genes.
If you find the web server useful for your work, then please cite:
P. Menzel, P. F. Stadler and J. Gorodkin: maxAlike: Maximum-likelihood based sequence reconstruction with application to improved primer design for unknown sequences, Bioinformatics, 2011, 27, 317-325.
P. Menzel, J. Gorodkin and P. F. Stadler: Maximum Likelihood Estimation of Weight Matrices for Targeted Homology Search, Proceedings of the German Conference on Bioinformatics 2009, LNI. 2009; 211-220 (PDF).
Input
Multiple Sequence Alignment
The first input file is a multiple sequence alignment of the already known homologous sequences. Supported file formats are Stockholm, CLUSTAL, or FASTA.
Example for Stockholm format
# STOCKHOLM 1.0 Dmel ATATTCTGCCGTCATAATGTAATAGTACGAATGTCTTGTGTTG Dsim ATATTCTGCCGTCATAATGTAATAGGACGAATGTCCTGTGTTG Dsec ATATTCTGCCGTCATAATGTAATAGGACGAATGTCCTGTGTTG Dyak ACATTCTGCCGGCATAATGCAATAAGACGAAAGTCCTGTGTTG Dere ATATTCTTCTTTCATAATGTAATAGGACGAATGTCCTGAGTGG Dana ATATTTTGCCGTCATAATGTAATAGGGCGAATGTTTTTTACAG Dpse ATATTATGCCGTCATAATATAATAGGCCAAACGTTTTTTGCAG Dper ATATTATGCCGTCATAATGTAATAGGCCAAACGTTTTTTGCAG Dwil ATATTTTGCCGTCATAATTCAATATGACAGTTTTTTTTTGGAT Dvir ACGTTTTGCTGTCATAATGATATAAATTGACGGCTCTTTGGGT Dgri ACGTTTTGCTGTCATAATGTAATAAATTGACGGTTTTTTGCGT //
Example for CLUSTAL format
CLUSTAL W (1.82) multiple sequence alignment Dmel ATATTCTGCCGTCATAATGTAATAGTACGAATGTCTTGTGTTG Dsim ATATTCTGCCGTCATAATGTAATAGGACGAATGTCCTGTGTTG Dsec ATATTCTGCCGTCATAATGTAATAGGACGAATGTCCTGTGTTG Dyak ACATTCTGCCGGCATAATGCAATAAGACGAAAGTCCTGTGTTG Dere ATATTCTTCTTTCATAATGTAATAGGACGAATGTCCTGAGTGG Dana ATATTTTGCCGTCATAATGTAATAGGGCGAATGTTTTTTACAG Dpse ATATTATGCCGTCATAATATAATAGGCCAAACGTTTTTTGCAG Dper ATATTATGCCGTCATAATGTAATAGGCCAAACGTTTTTTGCAG Dwil ATATTTTGCCGTCATAATTCAATATGACAGTTTTTTTTTGGAT Dvir ACGTTTTGCTGTCATAATGATATAAATTGACGGCTCTTTGGGT Dgri ACGTTTTGCTGTCATAATGTAATAAATTGACGGTTTTTTGCGT
Example for FASTA format
>Dmel ATATTCTGCCGTCATAATGTAATAGTACGAATGTCTTGTGTTG >Dsim ATATTCTGCCGTCATAATGTAATAGGACGAATGTCCTGTGTTG >Dsec ATATTCTGCCGTCATAATGTAATAGGACGAATGTCCTGTGTTG >Dyak ACATTCTGCCGGCATAATGCAATAAGACGAAAGTCCTGTGTTG >Dere ATATTCTTCTTTCATAATGTAATAGGACGAATGTCCTGAGTGG >Dana ATATTTTGCCGTCATAATGTAATAGGGCGAATGTTTTTTACAG >Dpse ATATTATGCCGTCATAATATAATAGGCCAAACGTTTTTTGCAG >Dper ATATTATGCCGTCATAATGTAATAGGCCAAACGTTTTTTGCAG >Dwil ATATTTTGCCGTCATAATTCAATATGACAGTTTTTTTTTGGAT >Dvir ACGTTTTGCTGTCATAATGATATAAATTGACGGCTCTTTGGGT >Dgri ACGTTTTGCTGTCATAATGTAATAAATTGACGGTTTTTTGCGT
RNA secondary structure annotation can be supplied in the Stockholm format by using a line starting with "#=GC SS_cons" followed by the secondary structure in Dot-Bracket Notation. Pseudoknots can be denoted by using upper-case and lower-case letters for the 5' and 3' part of a stem.
Example for Stockholm notation of RNA secondary structure with pseudo-knot
# STOCKHOLM 1.0 Species_name AACCGGAAAUAACCGGAAUAUGGGCCCCUGUUUUCCCGCGCACAGCACA .... #=GC SS_cons ..((((......)))).....(((...AAAA...)))....aaaa.... //
Tree
Together with the sequence alignment, a phylogenetic tree is needed, which contains at least some of the species that are also contained in the sequence alignment. These species will be used to infer the nucleotide probabilities in the target species (see below). The tree must be in Newick format, which is described here. All branches in the tree must be associated with distances and node names must be the same as the sequence names in the alignment file.
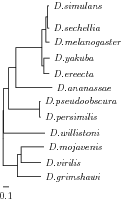
Example
Tree with 12 Drosophila species.(((((((Dsim:0.021,Dsec:0.024):0.029,Dmel:0.06):0.067, (Dyak:0.097,Dere:0.089):0.032):0.437,Dana:0.608):0.119, (Dpse:0.010,Dper:0.018):0.519):0.083,Dwil:0.691):0.013, ((Dmoj:0.382,Dvir:0.334):0.063,Dgri:0.395):0.241);
Sample trees
Sample species trees from several sources.
- Multiz 44 way from here.
- ENCODE 36 species from here.
- 12 Drosophila species from here.
Software for tree construction
Many software tools and web servers exist for estimating trees from existing multiple sequence alignments, e.g.:
- PHYML web server
- FastTree Download
- Exhaustive list of phylogenetic software and web servers by Joe Felsenstein
Name of Target Species
At last, you have to specify the name of the target species, the name must be contained in the tree file.
Example
In the example alignment D. mojavenis is missing, so we would specify Dmoj as the target species for the sequence reconstruction.
Window
It is possible to specify a start and end column of the alignment and the computation will only be done between (and including) these columns. The first column has number zero.
Consensus sequence probability threshold
After the computation is finished, two consensus sequences will be generated from the calculated nucleotide probabilities. In the first sequence, the nucleotide with the highest probability will be chosen at each site. In the second sequence, the probabilities must exceed the specified threshold to be part of the consensus sequence, otherwise it will contain an "N" at this position.
Highly informative sites
From the calculated nucleotide probabilities, it is possible to find windows which mostly contain highly probable predicted sites. You can specify the minimum length and the minimum average information content for each of these windows.
Output
Nucleotide probabilities
The main output file contains one line for each alignment column. The first field contains the column number. The second field contains the column number of the pairing partner of this column, if it had been specified in the #=GC SS_cons line in the alignment, otherwise it will be -1 for unpaired bases. The max_mu column yields the evolutionary rate (\mu), that maximizes the likelihood of the phylogenetic tree given the nucleotides from the alignment. The IC column contains the information content, calculated from the nucleotide probabilities. Then there are four columns containing probabilities for each base.
Example
These are the first lines of the output file, produced by the above sample alignment and phylogenetic tree for target species Dmoj. The first line shows that 100% probability is given to nucleotide A, since all sequences in the alignment contain this nucleotide. For column 2, C gets the highest probability with 0.67, etc. An information content of 2 is only assigned to columns with a nucleotide having probability 1.0.0 pair=-1 max_mu=0 IC=2 A=1 G=0 C=0 T=0 1 pair=-1 max_mu=0.892942 IC=0.508227 A=0.0705022 G=0.0534275 C=0.556843 T=0.319228 2 pair=-1 max_mu=0.308984 IC=1.10291 A=0.136081 G=0.812947 C=0.0143873 T=0.0365848 3 pair=-1 max_mu=0 IC=2 A=0 G=0 C=0 T=1 4 pair=-1 max_mu=0 IC=2 A=0 G=0 C=0 T=1 5 pair=-1 max_mu=0.873083 IC=0.859758 A=0.0701006 G=0.0527489 C=0.111391 T=0.765759 6 pair=-1 max_mu=0 IC=2 A=0 G=0 C=0 T=1 7 pair=-1 max_mu=0.234445 IC=1.41535 A=0.0577378 G=0.903541 C=0.0109292 T=0.0277915 8 pair=-1 max_mu=0 IC=2 A=0 G=0 C=1 T=0 9 pair=-1 max_mu=0.508516 IC=0.885656 A=0.0427397 G=0.0323887 C=0.178954 T=0.745918 10 pair=-1 max_mu=0.234445 IC=1.41535 A=0.0577378 G=0.903541 C=0.0109292 T=0.0277915
Sequence Logo
Additionally, a sequence logo is generated from the nucleotide probabilities. In brief, each sequence position is represented by a stack of the four letters A, C, G, and T. The higher the stack is in total, the higher the sequence conservation is at this site. The higher a individual letter is, the higher is his probability of occurrence. Thus, the sequence logo gives a intuitive feeling about the degree of conservation and which nucleotide to expect at each position. Read more about sequence logos here
Example
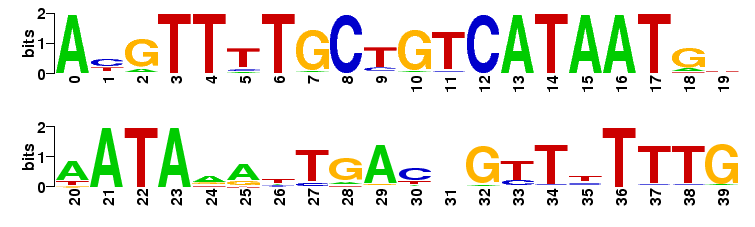
The sequence logos are created with the weblogo software.
Consensus sequences
From the nucleotide probabilities, two consensus sequences are computed. One sequence only considers nucleotides with probabilities about a probability threshold, which was set during submission. Positions with probabilities below this cutoff will be denoted as "N". The second sequence contains the highest probable nucleotide at each position.
Example
Predicted consensus sequence, with a probability cutoff 0.5 at each site:
>Dmoj predicted consensus sequence with probability cutoff 0.5
ACGTTTTGCTGTCATAATGNAATAAATTGACNGTTNTTTG
Predicted consensus sequence, without cutoff:
>Dmoj predicted consensus sequence
ACGTTTTGCTGTCATAATGTAATAAATTGACTGTTTTTTGTGT
Conserved Elements
Based on the computed nucleotide probabilities, the algorithm extracts those windows of certain length and a minimum average information content, that match the input parameters from the submission page.
Example
