
Welcome to the deepBlockAlign web page
Introduction
Many transcripts in cell undergo post-transcriptional processes that generate short RNA sequence fragments. When these fragments are mapped back to the reference genome they often form distinctive patterns that convey information on structure and processing of their parent transcripts. A well known pattern arises is formed e.g. by the miR and miR* reads generated from microRNA precursors.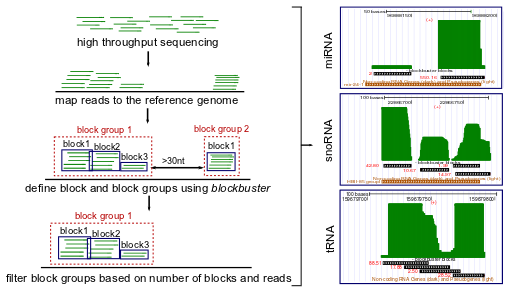
deepBlockAlign is a tool for the alignment of two RNA-seq read patterns. The analysis starts from closely spaced blocks of mapped reads, which we call block groups or read profiles. The comparison of two block groups is done in two stages.
- First, pairs of individual read blocks are compared using a modified Smith-Waterman alignment algorithm.
- Second, block groups are compared by means of a scoring function that evaluates both the similarity of the individual blocks and the pattern of distances within the block groups. For this purpose a modified sankoff algorithm is used.