Website usage
This website presents the comprehensive information related to the RNA structure predictions in 202 cyanobacteria genomes. The website offers the following main sections (see entries in the blue navigation bar on the top of the website):
CRS-Cyanobacteria
The landing page briefly describes the study, contains links to the main computational tools, the citation reference, a table with an overview over the number of predicted conserved RNA structures (CRSs), and a number of flat files for download of the main study results.
Browse CRSs
The browser page allows a user to filter the predicted CRSs by taxonomy (search area in the top right table), scores and pathway associations (check boxes in the top left). All CRSs that fit the user selection are shown in the table below. The main result table also has a search field that allows the user to search for gene names, ortholog identifiers, or CRS identifiers. A click on the header of each table allows to re-arrange the rows by the score of the clicked column.
Hoovering over the red information circles shows additional information to each respective entry.
Clicking on the blue underlined CRS identifier in the lower table navigates the user to details of the corresponding CRS in the next website section.
CRS details
This section shows all details associated with a CRS that a user selected either in the prior browser section or from the drop down box. The shown information includes all scores, potential pathway associations from the orthologous gene adjacent to which the CRS was predicted, the genomic loci of all conserved sequences in the CRS alignment, an overview over the species in which the CRS is predicted, and predicted transcriptional activity.
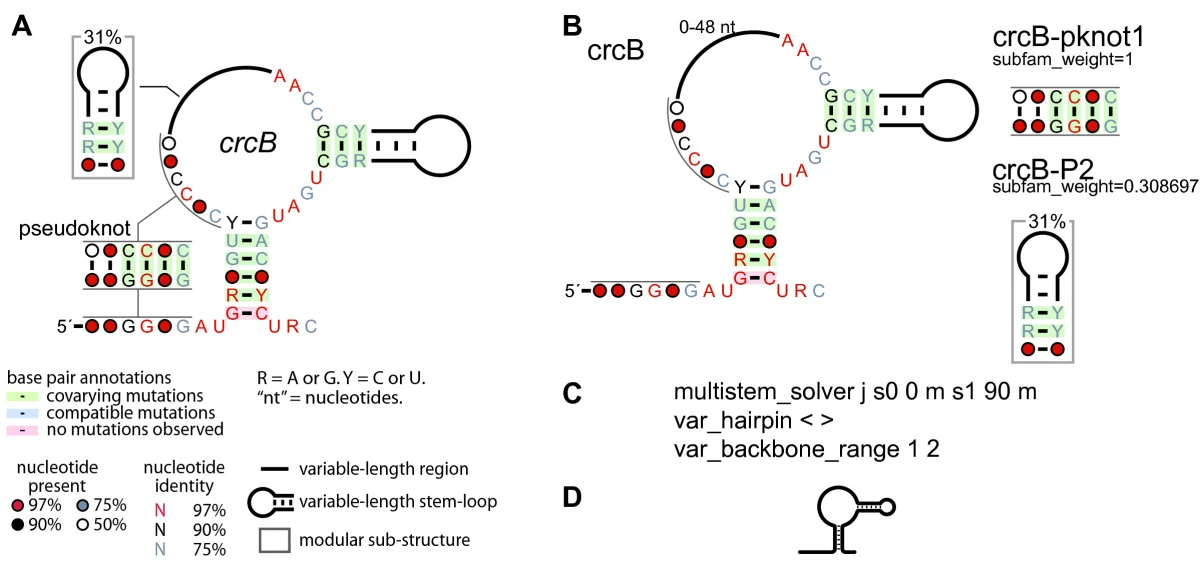
The details page also shows the post-processed conserved RNA structure with significant covarying basepairs highlighted in green (as calculated by R-scape). The structure figures were generated with R2R. The legend to these structures is as follows (Figure 1 in R2R publication):
CRS alignments
This sections shows the post-processed alignments of the novel CRSs.
The following post-processing steps were performed to improve the structure alignment and consensus structure:
- predict the consensus secondary structure from the CMfinder built alignment with PETfold (version 2.2)
- rebuild the covariance model (CM) with Infernal cmbuild (version 1.1.4) by using the CMfinder built alignment and the PETfold predicted structure
- realign sequences with Infernal cmalign to the new CM, and
- remove sequences with less than 65% canonical base pairs in the consensus structure.
The nucleotide positions are colored according to the JalView default scheme, that is adenine in green, cytosine in orange, guanine in red, and uracil in blue. The sequence identifiers on the left side are dot separated combination of the NCBI taxonomy ID, the BioSample identifier, and the Genbank Accession of the respective plasmid. The conserved RNA structure is shown below the alignment in dot-bracket notation.
Further, the filtered alignment is shown for improved and more compact visualization.
Species conservation
This section shows for each taxonomic order the fraction of studied species (177 in total) that the CRS anchoring orthologous gene is conserved in (barplot on the left side), and the fraction of species that the CRS is conserved in (barplot on the right side). The phylogentic tree of the group of orthologous genes in newick format can be downloaded (link at the bottom of the page). In this file the leaves in the tree are dot separated NCBI tax id + RefSeq chromosome accession + proGenomes gene ID (mostly RefSeq locus tag).
Known structures
The novelty of CRSs was assessed to known RNA structures from the Rfam database and a model for Rho-independent bacterial terminators (RNIE terminator). The known structures were annotated via a homology search with Infernal cmsearch for all 202 cyanobacteria genomes. The full annotation list of Rfam families and terminator sequences in the 202 genomes can be downloaded on the landing page (CRS-Cyanobacteria). This website section lists all hits for bacterial Rfam families and RNIE terminators that overlapped the search regions in which this study screened for RNA structures. A user can interact with this website section in a similar manner as in the CRS browser.
Use case
In this use case we want to explore the regulatory features of the ATP synthase of our favorite cyanobacteria Synechococcus sp. PCC 7002. Transcriptional and translational regulation of ATP synthesis in cyanobacteria is a complex process primarily centered on controlling the activity of ATP synthase, the enzyme responsible for ATP production. It is known that the small protein AtpΘ acts as a post-translational inhibitor of the ATP synthase complex. The half-life of the atpT transcript (KO identifier K01999) is dramatically different depending on the cell’s energy state ensuring that AtpΘ is produced only when needed (in the dark) and its synthesis is shut down rapidly when the cell’s energy state improves (in the light). This fast and efficient control mechanism is crucial for the cell’s ability to adapt to rapid light-dark cycles.
In addition, it is likely that sRNAs and other ncRNAs are part of the larger regulatory network that indirectly affects ATP synthesis by controlling the expression of other metabolic genes. To explore the conserved RNA structures that might be involved in the ATP synthesis in PCC 7002 we do the following:
At first, in the Browse CRSs section we filter the Ortholog to “K01999”. This gives us zero results telling us that no conserved structure has been predicted adjacent to atpT. Next, we filter the Phylogeny to “Synechococcales PCC 7002”, and the Pathway to “Photosynthesis”. This selection results in 1 of 409 predicted RNA structures. The CRS is listed in the bottom of the page with some of its features. By clicking on the name of the CRS K02110_downstream.h1_5 we enter its CRS details section.
In the CRS details section we can explore different features of the genomic location and conservation of the CRS, the RNA structure, and the transcriptional activity in selected cyanobacteria. CRS K02110_downstream.h1_5 has been found in 32 species, primarily in the orders Chroococcales and Nostocales, and some species in Oscillatoriales and Synechococcales. By following the link View species conservation of the CRS anchoring orthologous gene we can see in the bar plot on the left side that the search region (an intergenic region of at least 20 nts downstream of K02110) exists in almost all investigated species. From here we can take the link View CRS details to go back. In the CRS details section we see that the location of the CRS in Synechococcales sp. PCC 7002 is CP000951:768,074-768,129 on the reverse strand. We can also get more info about the anchoring orthologous gene (ATPF0C, atpE; F-type H+-transporting ATPase subunit c) from the KEGG database, for instance to explore the genomic locus, by clicking K02110. At the KEGG orthology webpage we click on Taxonomy, on the KEGG Taxonomy Mapping webpage we search our genome (i.e., “PCC 7002”) and follow the link (sys), on the KEGG genome webpage we search our gene again (i.e., “K02110”) and select in the search results SYNPCC7002_A0738, and finally on the KEGG locus tag webpage we click on Genome browser. Now we can identify the synteny of the search region and the anchoring gene atpE which is on the reverse strand and consists of the upstream adjacent genes atp1 and atpB, and the downstream adjacent genes atpG, atpF, atpH, atpA, and atpG. All these genes are located on the same transcriptional unit (operon). Back in the CRS details section we can also get more info about the associated pathways from the KEGG database by clicking the pathway of interest, here map00195 Photosynthesis. Exploring the conserved structure we see that it is a long hairpin loop without any interior loops, little sequence conservation (average sequence identity of 50.2%), but 14 observed covarying basepairs (R-scape; green highlighted basepairs in the R2R structure image). By clicking View alignment we can explore the predicted structure alignment in more detail. The post-processed alignment at the top has still many gap regions, whereas the filtered alignment in the bottom is more compressed by having removed sequences with nucleotides in an alignment column with more than 95% of gaps (Note: this resulted in a filtered alignment with new sequences that have sequences with nucleotides in an alignment column with more than 95% of gaps). For further analysis, the alignments can be downloaded in STOCKHOLM or FASTA format, and the covariance model built on the post-processed alignment can be downloaded as well for search in other genomes.
We should also check if any annotated RNA structures can be identified that are by their genomic origin associated to the ATP synthesis in PCC 7002. In the Known structures section we filter the Phylogeny to “Synechococcales sp. PCC 7002” resulting in 0 known structures. Looking more broadly in evolution, we filter the Phylogeny to “Synechococcales” which results in 80 of 177 species in our screen, and 51 of 3981 genomic locations of Rfam structures, and 0 of 42069 transcription (RNIE) terminators. Scrolling through the list of known structures we see that all hits are from the same RNA family RF01067 which is the ATPC RNA motif and was found in different Prochlorococcus species and Synechococcus species, however, none of the species closely related to PCC 7002. We follow the link RF01067 to the Rfam page and learn that the ATPC RNA motif is a conserved RNA structure found in certain cyanobacteria. It is apparently ubiquitous in Prochlorococcus marinus, and is present in many species in the genus Synechococcus. The RNA is always found within an operon encoding subunits of ATP synthase, and it is always located downstream of the gene encoding the A subunit of ATP synthase, and upstream of the C subunit gene. This location is consistent with a cis-regulatory element, but also with a non-coding RNA that is transcribed with the ATP synthase genes. In addition, the Rfam page states that simple RNA structures called stem-loops have been reported in the ATP synthase operons of various cyanobacteria, but not structures such as the 3-stem junction that is the main feature of the ATPC RNA motif. It is likely that the mentioned stem-loops co-localize with our CRS candidate K02110_downstream.h1_5 that we have shown being conserved in different cyanobacterial orders including our species of interest.