|
|
|
|
|
IntroductionContents:1. The editorThe SARSE editor is implemented in Java and with a graphical front-end in Swing and is described in detail in
The editor comes with a variety of functionalities, which are briefly described here. The editor can read alignments in widetext format, column format (http://genome.ku.dk/resources/colformat) and fasta format all formats that are used in the RNA databases SRPDB and tmRDB (Andersen et al., 2006). SARSE has the ability of a split view (Figure 1A) so the left and right part of a stem separated in the sequence can be viewed and edited simultaneous. Selection of base pairs works like this: when clicking on a single or group of base pairs the complement bases can be highlighted as well and in split view shown in the other window right away (Figure 1A). This enables the user to readily identify complement bases and carry out an operation on them. Operations can be basic edit functions such as changing what bases pairs (for example in a block of selected nucleotides), and these will be base paired according to the pairingmask. The pairingmask itself can also be edited. Another basic function is the movement of smaller parts of sequences.
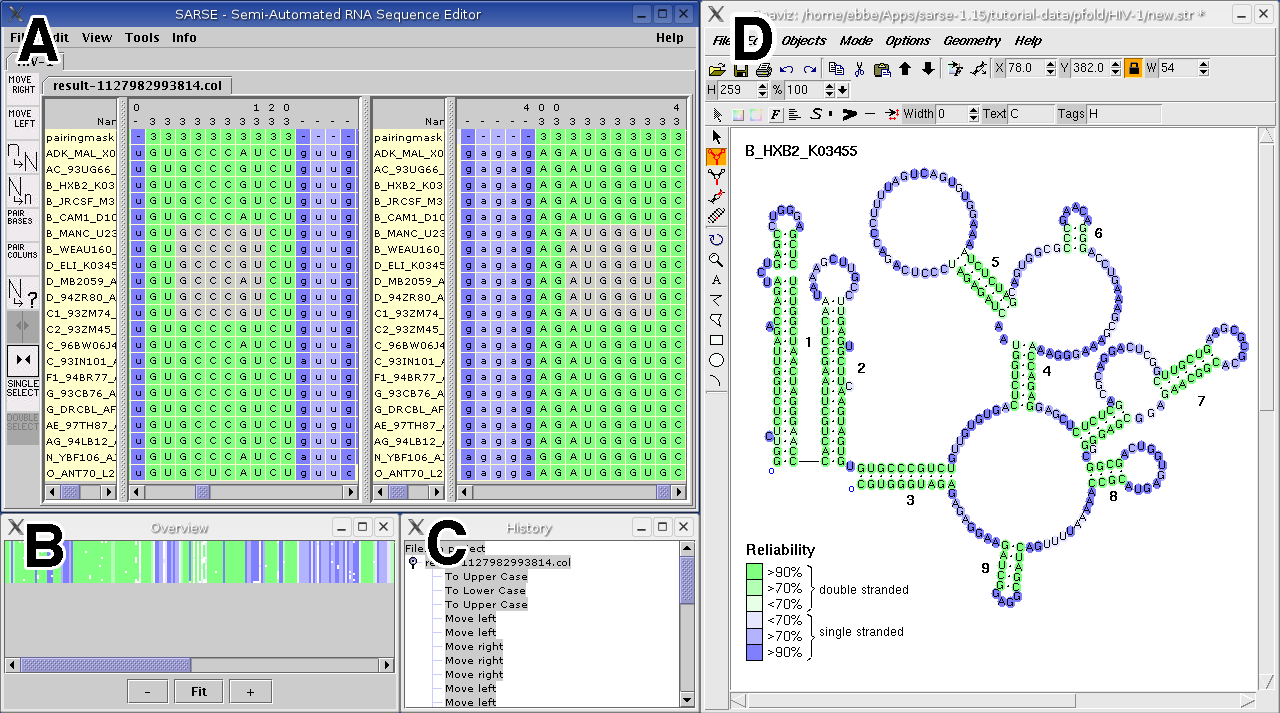
Figure 1: Suggested work screen for full functionality. (A) The SARSE editor window is shown in split view of an alignment that has been analyzed by Pfold. The colors correspond to the assigned reliability. (B) the Overview window. (C) The History window. (D) The alignment is imported in Rnaviz and the appropriate colors are added as background. A reliability color scale is shown. The full alignment can be overviewed in the Overview window (Figure 1B). This window displays a representation of the alignment colors, and allows interactive browsing of the alignment, since clicking in the Overview window will shift the Editor window to the corresponding position in the alignment. To keep track of the basic editing SARSE has a history function that makes it possible to view the changes that has been made, the history is saved with the project and thus accessible across sessions. The history function (Figure 1C) also allows for unlimited undo/redo actions also across sessions. To visualize the secondary structure of the alignment we recommend to use one of the available secondary structure editors e.g. RnaViz 2 (De Rijk et al., 2003). Format converters of the RNAdbtools package allows the alignment file to be opened in RnaViz with the same colors as in SARSE (Figure 1D). 2. The programs toolboxIn the programs toolbox it is possible to run a selected collection of programs from rnadbtools or other accessible programs. As computational tools are continuously developed SARSE enable the user to add his/her own programs for analyzing structural alignments. After executing the programs the results can be loaded into the editor and the positions are colored e.g. according to whether a series of base pairs are inconsistent according to the pairing mask or a stem can be extended in some of the sequences. 
Figure 2: The Programs window. The left panel shows the Programs Packages. Clicking on one of the packages will show the programs contained in the package to the right. Likewise, when executing pfold a new pairingmask can be loaded into the editor replacing the old one. Throughout all operations a directory is associated with the project and all data such as dotplots generated by the external programs can be accessed in that directory. In this way the editor maintain the flexibility in running other programs by making it possible to access the type of output they generate by nature. 3. RNAdbtoolsThe rnadbtool package of programs was developed to allow the handling of RNA alignments, update and analysis (Gorodkin et al., 2001). The homepage can be found here (http://genome.ku.dk/resources/rnadbtool). The rnadbtools are based on the column format (http://genome.ku.dk/resources/colformat). 4. PfoldThe Pfold algorithm predicts the common secondary structure for an alignment of sequences (Knudsen and Hein, 1999, 2003). It uses a Stochastic Context-Free Grammar (SCFG) and evolutionary history as the basis of prediction. 5. PclusterThe Pcluster method is introduced here:
In a multiple RNA alignment, there may be sequences that disrupt the Pfold secondary structure prediction for two reasons: poor alignment and variations in structure. This problem may in part be solved by splitting the sequences into smaller groups for which both the structures and the alignments are consistent. Doing this, alignment errors may become apparent and can be corrected. The Pcluster method clusters RNA sequences into such groups according to structure without performing new alignments. 6. FoldalignMFoldalignM is used for aligning sequences based on RNA secondary structure (Torarinsson et al., 2007). 7. References
Comments, questions, etc., email
webmaster@sarse.ku.dk. |
| Last updated November 5th, 2007 by E. S. Andersen, A. Lind-Thomsen and J. Gorodkin |