
CRISPRon-BE(v1.1): CRISPR-Cas9 base editing guide design
Help
The CRISPRon-BE webserver is dedicated to the design of guide RNAs (gRNAs) for CRISPR/Cas9-derived base editing experiments. Currently, our webserver supports two types of base editor predictions: adenine base editors (ABE, converting A:T base pairs to G:C base pairs) and cytosine base editor (CBE, converting C:G base pairs to T:A base pairs). Specially, the model designed for ABE (CRISPRon-ABE) is trained on data from ABE7.10 and ABE8e, while the model for CBE (CRISPRon-CBE) is trained on data from BE4.
The CRISPRon-BE model can predict the nucleotide conversion efficiency induced by designed gRNAs using base editors, as well as the detailed frequency for each potential edited product. If you are interested in inducing a specific nucleotide conversion or correcting a point mutation at a particular genomic region, simply input your target DNA sequence along with the specific nucleotide of interest into our CRISPRon-BE webpage. In approximately one minute, you will receive all the potential designed gRNAs for the given DNA sequence and their corresponding edited products.
In particular, considering that the base editing from different deaminases and experimental platforms are diverse, CRISPRon-BE allows users for a tailormade prediction depending on the dataset most like which datasets used for training. See detailed tailormade strategies in below section Select dataset weights for your prediction.
The webserver can be used with three different kinds of input: a custome input sequence or a genomic range or a gene name. For the first case, the sequence can be any DNA sequence, mapping or not mapping the the target genome, while for the later two cases the target genome must be specific.
Format for INPUT-1: Targeting a custom input sequence
Paste-in a target DNA sequence in plain or in fasta format as shown in the examples below and press submit.
If your sequence is from one of the organisms listed in the drop down menu, it is recommended to select
that organism in the form, even if your input sequence doesn't map
perfectly to the target genome.
Example of sequence in plain:
AGGCAACCTGAGGACTTGTCTGAGGATGGGGCCGCAA
Example of sequence in fasta format:
>myseq_id AGGCAACCTGAGGACTTGTCTGAGGATGGGGCCGCAA
Hint: how to get a DNA sequence. A possible way to obtain the DNA sequence of a given region or gene is to search for it in the UCSC genome browser and then use View->DNA. If the region contains a mutation in your subject, edit the sequence accordingly before submitting it to CRISPRon-BE (see "Examples of input/output" below).
Input criteria. The input must contain only the following five canonical bases 'A,a', 'C,c', 'G,g', 'T,t' and 'U,u' or unknown bases 'N,n'. Targets that include unknown bases are omitted from the output. Predictions are made for a possible target site only if at least 30 nt made of 4 nt + target (20 nt) + PAM (NGG) + 3 nt are present in the given sequence.
Format for INPUT-2: Targeting a gene
First, select one of the listed genomes in the drop down menu. Then, select a gene in the form by typing its name (a drop-down menu will appear after 3 characters), and press submit. In this mode, only exons (coding exons or UTRs) will be considered as targets. If you wish to target introns you must use one of the other input options. If either the genome or the gene is not listed, you should use the "Format for INPUT-1" and manually enter your target sequence as explained above. In addition to targets in exons, targets in the 300 nt sequence up-stream and down-stream of each transcript are also shown in the output. Input example:
SLC10A4
Format for INPUT-3: Targeting a genomic region
First, select one of the listed genomes in the drop down menu. If the genome you are interested in is not listed, please use the "Format for INPUT-1" described above. Then, type in a genomic region in the dedicated form, and press submit. The genomic region should be specified in the following format: chrN:start-end, where chrN is the identifier of the chromosome, and start/end mark the genomic range, as in the example below.
chr5:73,164,226-73,170,298
Select dataset weights for your prediction
Considering the diversity of base editing from different deaminases and different platforms for concreate editing, we proposed a fused model by using a combined dataset and flagging the data sources to train the model. In this way, prediction on datasets benefits from the others, while allowing new predictions based on a weighted combination of the datasets used for training.
Currently, five datasets are used for CRISPRon-ABE training, including SURRO-seq (Xiang et, al., 2021, Pan et, al., 2022), Song (Song et al., 2020), Arbab (Arbab et al., 2020), Kissling ABEmax and Kissling ABE8e (Kissling et. al., 2025) datasets and three datasets are used for CRISPRon-CBE training, including SURRO-seq (Xiang et, al., 2021, Pan et, al., 2022), Song (Song et al., 2020) and Arbab (Arbab et al., 2020) datasets. These datasets are diverse in terms of gRNA editing efficiencies and outcome frequencies.
Overview of diverse datasets applied in CRISPRon-BE
| Editor | Dataset | Deaminase | sgRNA | gRNA targeted sites |
|---|---|---|---|---|
| ABE | SURRO-seq | ABE7.10 | standard | genomic sequence |
| Song | ABE7.10 | standard | genomic sequence | |
| Arbab | ABE7.10 | modified* | synthetic sequence | |
| Kissling ABE7.10 | ABE7.10 | modified | pathogenic and monogenic sequence | |
| Kissling ABE8e | ABE8e | modified | pathogenic and monogenic sequence | |
| CBE | SURRO-seq | BE4 | standard | genomic sequence |
| Song | BE4 | standard | genomic sequence | |
| Arbab | BE4 | modified | synthetic sequence |
* Modified sgRNA with extended stem and modified nucleotide (Phillips-Cremins et,al.,2013)
When predicting on new gRNAs, users can perform tailormade predictions by assigning weights to training sets that are most similar to the new gRNAs. By clicking "Advanced", users can select the training sets most similar to their own data for a customized prediction.
We also provide default selection for each base editor.
- ABE7.10: selection of SURRO-seq and Song dataset since these two datasets applied a standard scaffold sequence in their experiments
- ABE8e: selection of Kissling ABE8e, the only ABE8e data for training the model
- BE4: selection of SURRO-seq and Song dataset since these two datasets applied a standard scaffold sequence in their experiments
A more advanced option for weight assignment is available through the command line — for example, assigning four times the weight to the SURRO-seq dataset compared to the Song dataset.
Examples of CRISPRon-BE for gRNA design
Here we use one example to show how to use CRISPRon-BE for gRNA design.
One SNP NM_004006.3(DMD):c.5804G>A (p.Gly1935Asp) is collected in the ClinVar Database, which represents a point mutation at chromosome X: 32342218, changing from “C” to “T” on the annotated (+) strand of the reference genome (hg38). To correct this SNP using a base editor, you can design a gRNA complementary to the annotated (-) strand. This allows the nucleotide "A" in the patient to be corrected to "G" by ABE.
1. Prepare for the target DNA sequence.
CRISPRon-BE requires the DNA sequence with a PAM motif ("NGG") for prediction. In this case, the target nucleotide “A” is in the complementary strand, so we extract the sequence from the complementary (-) strand for gRNA design. Considering that ABE or CBE has strong positional editing efficiency at positions 3 to 10 on the PAM-distal side (editing window, where base pair conversions by base editors mainly happen in this region), only SNPs located within this editing window can be corrected efficiently. Therefore, we extract the nucleotide sequence upstream by 13 nt and downstream by 23 nt for gRNA design.
For the target nucleotide located in the complementary (-) strand (5’ to 3’), the sequence from chrX:32342195-32342231 is extracted as follows:
AGGCAAGCTGAGGGCTTGTCTGAGGATGGGGCCGCAANext, manually change the 14th nucleotide from “G” to “A” to reflect the mutation:
AGGCAAGCTGAGGACTTGTCTGAGGATGGGGCCGCAA
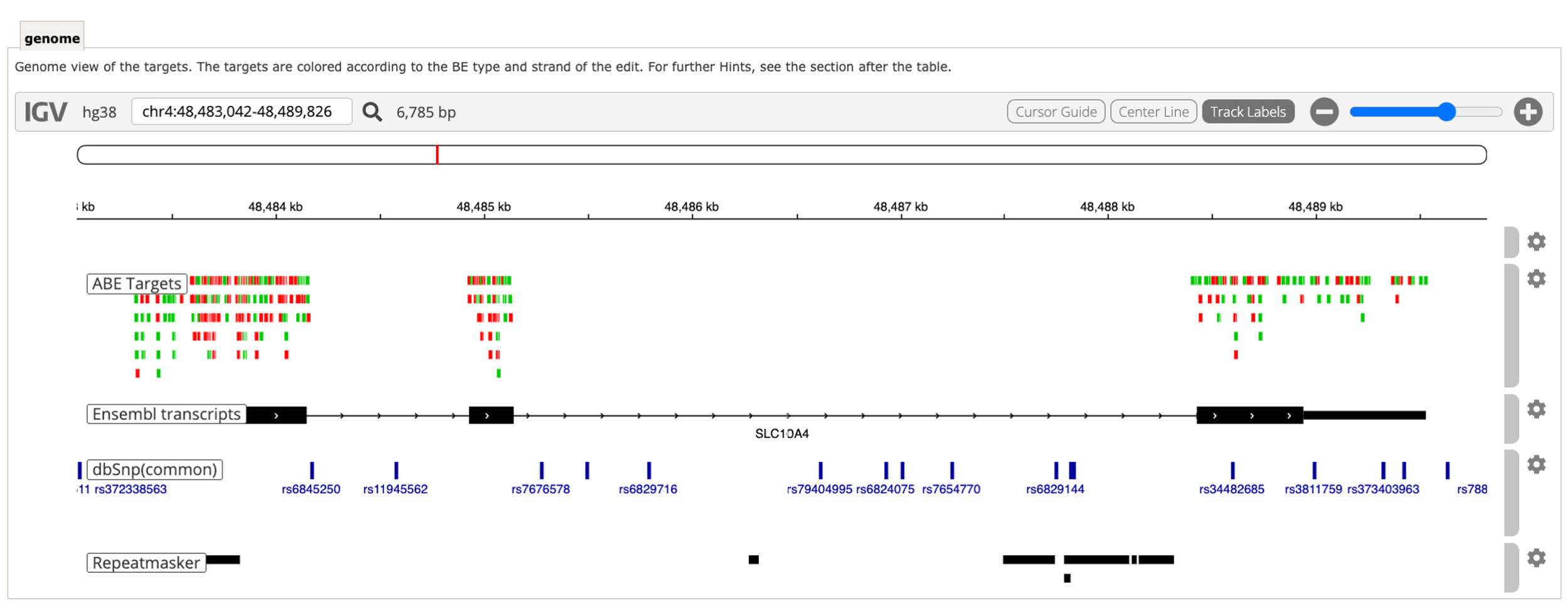
In CRISPRon-BE, the webserver allows to search potential gRNAs on both strands to make it convenient. In this case if you input the DNA sequence "TTGCGGCCCCATCCTCAGACAAGTCCTCAGCTTGCCT" which is reverse complementary of the input sequence mentioned above, you will obtain the same designed gRNA sequences. But in IGV, the rectangles represnting editing windows will appear in different colors and positions, as the gRNAs are located on different strands compared to the original input sequence.
2. Predict designed gRNA editing efficiency and outcome frequencies.
After pasting the sequence with the mutation into CRISPRon-BE, and choosing the suitable base editor (ABE7.10, ABE8e or BE4), we can generate potential designed gRNAs along with their predicted outcomes. Here we choose "ABE7.10" base editor as example to show the results. In IGV, there are three different gRNAs capable of correcting the SNP. Each rectangle in IGV represents the editing window for a potential gRNA. For instance, there are two "A"s in the editing window for the first gRNA. To correct the mutation, we target the second "A" (14th nucleotide) within the editing window, while ensuring high editing efficiency specifically at this position, and avoiding unintended editing of the first "A" (11th nucleotide) in the editing window. Clicking each rectangle (gRNA) in IGV shows a brief information of this gRNA, including the base editor type (BE_type), strand compared with input sequence (BE_strand), sequence in the editing window (editing_window), gRNA editing efficiency(total_gRNA_efficiency), the outcome sequence with highest predicted freqeuncy (main_outcome) its corresponding predicted frequency (outcome_frequency), and its location in the input sequence (Location).

In the table below, detailed predictions for gRNA editing efficiency and outcome frequency are provided. Hovering over the columns in the table, you can also get the introduction of the given column. In this table, only outcomes with frequency >= 1.0% are shown here. You can download the result to see the rest.
- id_be: ID and base editor type of potential gRNAs. Use this column to sort or search for specific gRNAs.
- coords: Location of the 30 nt target sequence (4 nt upstream + 20 nt protospacer + 3 nt PAM + 3 nt downstream) in the input sequence.
- grna: Sequences of potential 20 nt gRNAs.
- be-outcome: Potential outcomes from the designed gRNAs. Two types of outcomes are included: Wild-type outcome: Identical to the gRNA sequence. The editable nucleotides ("A" for ABE and "C" for CBE) are shown in uppercase and colored. Edited outcome: Edited nucleotides compared to the gRNA sequence are highlighted.
- frame: Six different reading frames are used to translate the corresponding protein sequences from the given outcome sequence. By default, only the translated protein sequence from the plus strand in frame 0 is displayed. To view protein sequences translated in different frames, click on "Select columns to show".
- total_eff: Predicted gRNA editing efficiency.
- outcome_freq: Predicted frequency of a specific outcome for each gRNA. For wild-type outcomes, the frequencies are showing "NaN" as we mainly focus on edited outcomes.
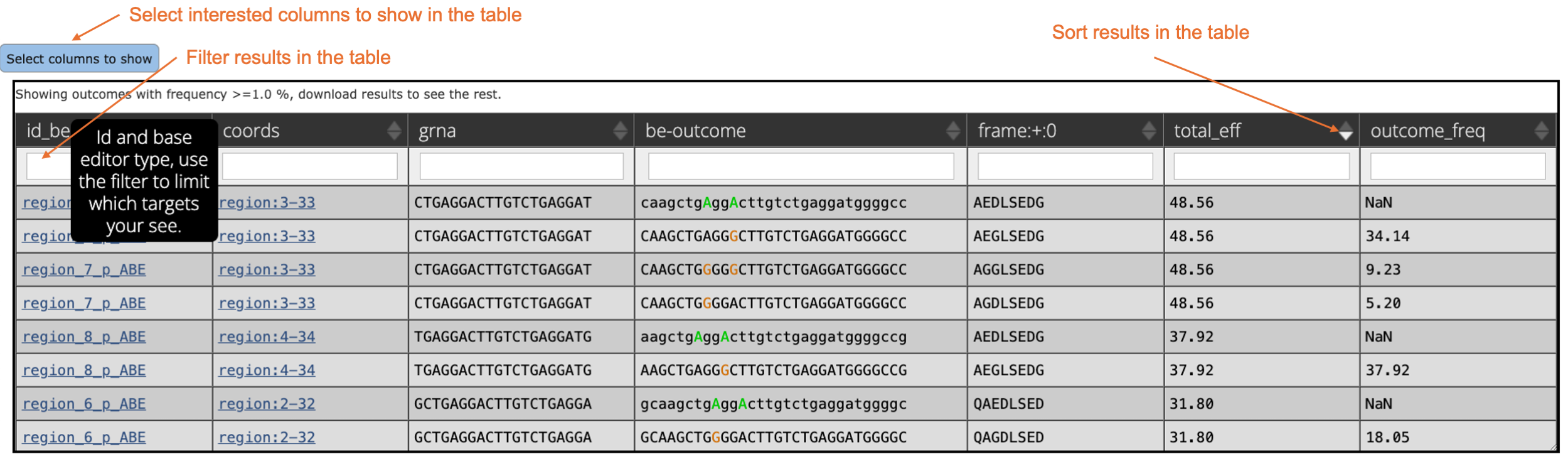
If you are interested in the edited result from one specific gRNA, you can click on the gRNA ID or filter the table using the gRNA sequence or gRNA ID.
In this example, three gRNAs sequence are available. “GCTGAGGACTTGTCTGAGGA”, “CTGAGGACTTGTCTGAGGAT” and “TGAGGACTTGTCTGAGGATG”. Their predicted gRNA editing efficiencies are “31.80”, “48.56” and “37.92”, respectively. Based solely on editing efficiency, the second gRNA is the highest at 48.56, making it the best choice, while the third gRNA at 37.92 is also considered acceptable.
Considering the bystander effects from the base editor, we also evaluated their frequencies of correcting this mutation without any bystander effects. The outcome frequencies for the gRNAs are 11.84, 34.14, and 37.92. The third gRNA stands out as the best option with the highest outcome frequency and minimal bystander edits (other edited outcomes with frequencies less than 1). In contrast, the second gRNA shows stronger bystander effects where both "A"s in the editing window can be edited simultaneously with a frequency of 9.23.
In this example, we recommend using the third gRNA "TGAGGACTTGTCTGAGGATG" to correct this mutation. It offers both high gRNA editing efficiency and minimal bystander effects, making it the optimal choice.
The example above demonstrates how to use CRISPRon-BE to design gRNAs for correcting mutations in the genome,
applicable to any DNA sequence as an input for gRNA design by base editors.
CRISPRon-BE also supports inducing mutations in the reference genome through a user-friendly interface.
Users can input DNA genomic locations or gene names to obtain results similar to inputting DNA sequences extracted from the reference genome.
In this scenario, DNA sequences from both strands are searched and the designed gRNAs are labeled with different colors in IGV for clarity.
If you are searching a coding gene, the resulting protein sequence for the canoical CDS is shown rather than the translation using six different frames.
References
Arbab M, Shen MW, Mok B, Wilson C, Matuszek Z, et al. 2020. Determinants of Base Editing Outcomes from Target Library Analysis and Machine Learning. Cell 182: 463-80 e30
Kissling L, Mollaysa A, Janjuha S, Mathis N, Marquart KF, et al. 2025. Predicting adenine base editing efficiencies in different cellular contexts by deep learning. Genome Biol 26: 115
Pan X, Qu K, Yuan H, Xiang X, Anthon C, et al. 2022. Massively targeted evaluation of therapeutic CRISPR off-targets in cells. Nat Commun 13: 4049
Phillips-Cremins JE, Sauria ME, Sanyal A, Gerasimova TI, Lajoie BR, et al. 2013. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell 153: 1281-95
Song M, Kim HK, Lee S, Kim Y, Seo SY, et al. 2020. Sequence-specific prediction of the efficiencies of adenine and cytosine base editors. Nat Biotechnol 38: 1037-43
Xiang X, Corsi GI, Anthon C, Qu K, Pan X, et al. 2021. Enhancing CRISPR-Cas9 gRNA efficiency prediction by data integration and deep learning. Nat Commun 12: 3238
Citing
If you are using the results of the webserver in your publication, please cite:
Deep learning models simultaneously trained on multiple datasets improve base-editing activity prediction
Sun Y, Qu K, Corsi GI, Anthon C, Pan X, Xiang X, Jensen LJ, Lin L, Luo Y*, Gorodkin J*
Nat Commun. 2025 Nov 7;16(1):9821
[ PubMed | Paper | Webserver | Software ]
Feedback
We greatly appreciate your comments. Open Feedback form in a new tab. Alternatively you can E-mail us with your questions and comments.