
mirDBA: microRNA discovery by similarity search to a database of RNA-seq profiles
Version
This is version 1.0.Input
The web server takes BED file as standard input thereby removing the need for any data pre-processing steps as would be required for such kind of analysis using the standalone version of deepBlockAlign. Besides, the user can adjust several optional parameters that affect prediction results, although the default setting is optimal in many cases.sample BED format
#chr start end id number of reads strand chr1 20229 20366 GSM34290_325* 50$ + chr1 20230 20369 GSM34290_328* 13$ +* id is comprised of an [identifier]_[number of times the read is mapped (optional)]
$ normalized read count (read count / number of times the read is mapped)
We decided to use input in BED format due to two main reasons:
- Conversion from BAM to BED considerably reduces the size of file that needs to be uploaded, for example this RNA-seq dataset of size 1.3 GB from ENCODE is reduced to 23 MB on conversion to BED format. Low file size eventually leads to faster upload and less load on the web server.
- We provide a non-dependency perl script, bam2bed.pl that
- convert BAM into BED format.
- format read id such that it provide information about multiple and uniquely mapped reads.
- normalize the read count based on mapping frequency.
Parameters
- Reference genome: reference genome for the input BED file. Required to annotate block group(s) processed from the input BED file. Currently, the web server provides non-coding RNA annotation for thirteen genome assemblies. However, web server can be used for reads mapped to genome assemblies for which the annotations are not available by choosing the 'other' option from the drop down box. Only difference would be that the processed block groups or read profiles will not be annotated in the results.
- Match score: given two read profiles, X and Y, match is the score for aligning two positions Xi and Yj, if the expression difference between Xi and Yj is lower Threshold (expression difference).
- Mismatch score: given two read profiles, X and Y, mismatch is the score for aligning two positions Xi and Yj, if the expression difference between Xi and Yj is higher than the Threshold (expression difference).
- Gap penalty (initialize): given two read profiles, X and Y, gap initialization is the score for aligning two positions Xi and Yj, when one of them is having a gap initialized.
- Gap penalty (extension): given two read profiles, X and Y, gap extension is the score for aligning two positions Xi and Yj, when one of them is having a gap extended.
- Threshold (expression difference): given two read profiles, X and Y, threshold is the expression difference between Xi and Yj to consider them as a match or as a mismatch.
- Distance weight: given two read profiles, X and Y having M and N blocks, distance weight is the weight to influence the distance between the blocks in X and Y. Higher the distance weight, less tolerant will be the algorithm to the difference in distance between the blocks in X and Y.
- Block similarity weight: given two read profiles, X and Y having M and N blocks, block weight is the weight to influence the alignment score between the blocks, Mi and Nj in X and Y, respectively. Higher the block weight, more weightage will be given to the similarity score between blocks Mi and Nj of block groups of X and Y, respectively.
- Gap penalty: given two read profiles, X and Y having M and N blocks, gap penalty is the score if a gap is inserted in the alignment due to dissimilar number of read blocks in X and Y.
- Threshold score: minimum alignment score between two read profiles to consider them as similar (default: 0.7).
- Maximum number of alignments: maximum number of alignments to report for each query block group or read profile (default: 2).
- Database: it is comprised of 2,540 miRNA read profiles or block groups derived from preprocessing of short RNA-seq data from miRBase.
Pre-processing of input BED file
The input BED files are preprocessed to- group overlapping and closely spaced reads into read blocks which themselves are grouped to form a block group or read profile using blockbuster.
- flag block groups or read profiles as annotated, if overlapping to a known annotation of ncRNA.
- create index of BED files in order to facilitate faster search.
- create index of block groups in order to facilitate faster search.
- create UCSC track files that are used to visualize read profiles in the UCSC genome browser.
A summary of preprocessing results are shown below

- Reads: total number of reads in the input BED file.
- Uniquely mapped reads: total number of reads that mapped at <=2 times in the genome.
- Tags: total number of tags (unique read sequences) in the input BED file.
- Uniquely mapped tags: total number of tags that mapped at <=2 times in the genome.
- miRNA: total number of block groups that overlapped with known miRNA annotations.
- snoRNA: total number of block groups that overlapped with known snoRNA annotations.
- tRNA: total number of block groups that overlapped with known tRNA annotations.
- other: total number of block groups that overlapped with known other ncRNA annotations.
- unannotated: total number of block groups that did not overlap with any known ncRNA annotations.
- total: total number of block groups that are processed from the input BED file.
Output
The alignment results are presented in a multi-page tabular format as shown below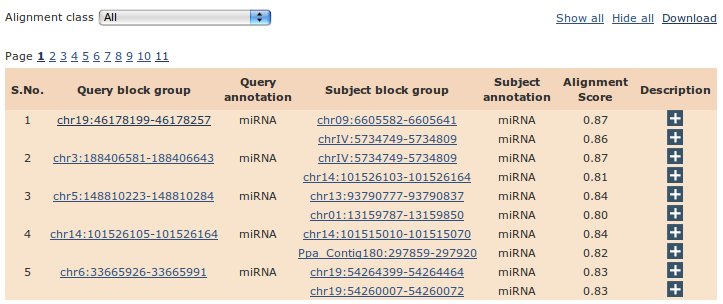
- Alignment class: To efficiently filter genomic regions based on their annotation status and entropy of read profiles, we have classified the alignment results into four classes
- unannotated (entropy<=2): query loci is unannotated and one of the read profile has entropy <=2.
- annotated (entropy<=2): query loci is annotated and one of the read profile has entropy <=2.
- unannotated (entropy>2): query loci is unannotated and none of the read profile has entropy <=2.
- annotated (entropy>2): query loci is annotated and none of the read profile has entropy <=2.
- Query block group: query a block group or read profile derived from the input BED file using blockbuster.
- Query annotation: non-coding RNA annotation of query block group.
- Subject block group: subject block group or read profile from the block group database (database search) prepared by preprocessing nine short RNA-seq datasets from ENCODE.
- Score: alignment score between two read profiles computed using deepBlockAlign. Score lies between 0 and 1, higher it is more similar are the two profiles.
- Description: complete description and graphical representation of the alignment between read profiles.

- name: non-coding RNA overlapping with the block group coordinates from the reference genome.
- coordinate: genomic coordinates of the read profile or block group. User can view the read profile at UCSC genome browser on clicking over this link.
- reads: total reads within the block group.
- tags: total tags (reads with unique coordinates) within the block group.
- blocks: total number of blocks within the block group defined during the preprocessing step of input BED file.
- entropy: entropy measures the randomness in the arrangement of reads within the block group. Small value indicate well-defined patterns, while large values arise from blurred patterns indicative of degradation product. More details are in the original paper on deepBlockAlign.
- ncRNA: non-coding RNA annotation, if the block group coordinates are overlapping to the coordinates of a known non-coding RNA in the reference genome.
- loci: genomic loci (exon, intron, 5' UTR or 3' UTR) overlapping to the coordinates of block group in the reference genome.
- unique reads (%): percentage of uniquely mapped reads within the block group.

- block group alignment: shows optimal alignment of blocks between two block groups. A block is composed of closely spaced set of reads within a block group and are defined during the preprocessing step.

- block alignment: shows the optimal position-wise alignment of all pair of blocks that aligned optimally during the block group alignment. The alignment is between read count of two blocks normalized with respect to the total read counts within the respective block groups. More details are in the original paper on deepBlockAlign.